Project Description
Valorizzazione delle colture da rinnovo in ambienti toscani in previsione dei futuri cambiamenti climatici
Durante il primo anno di progetto la Scuola Superiore Sant’Anna di Pisa (SSSA) si è focalizzata sulla ricerca bibliografica e sulla pianificazione delle attività previste per gli anni a seguire. Il progetto VARITOSCAN prevede l’intervento di SSSA nella caratterizzazione dettagliata dell’assetto genetico di individui di miglio e mais mediante utilizzo di marcatori molecolari di tipo SNPs (polimorfismi di singola base nucleotidica) analizzati con approcci di next-generation sequencing o tramite analisi con Array specifici.
Acciocché ciò avvenga con la massima efficienza, durante il primo anno di progetto il personale strutturato SSSA ha eseguito un’estensiva ricerca bibliografica sulla letteratura disponibile e riguardante la caratterizzazione molecolare di miglio e mais. Per il solo miglio, nel 2019 sono stati pubblicati ben 58 articoli scientifici su un totale di 813, in linea con un trend in costante crescita (Fig. 1). Il mais, che è una specie oggetto di ricerca da più tempo e studiata da una comunità scientifica più ampia, ha visto 4 470 articoli pubblicati nel 2019 (su un totale di 108 029).
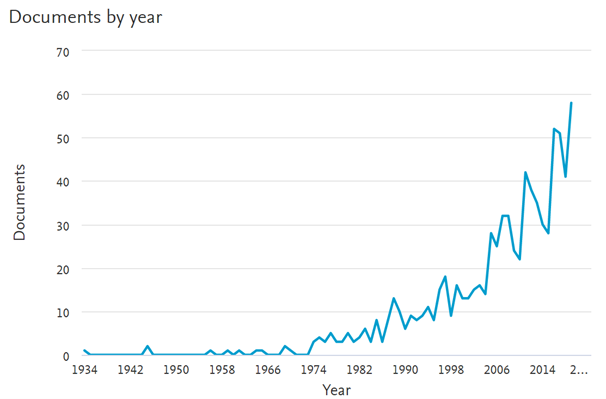
Fig. 1. Numero di articoli riguardanti la specie miglio (Panicum miliaceum) pubblicati ogni anno a partire dal 1934 (fonte: Scopus)
Da tale ricerca bibliografica sono state derivate strategie relative all’implementazione delle attività di progetto negli anni 2020-2021, in particolare riguardo alle tecniche di genotipizzazione da utilizzare. La strategia preferenziale per entrambe le specie è stata identificata nel double-digestion restriction site associated DNA markers (ddRAD), una tecnica affine al genotyping-by-sequencing (GBS) (Fig. 2), che permette la produzione di migliaia di SNPs distribuiti su tutto il genoma passando per una riduzione della complessità del genoma ad opera di enzimi di restrizione.
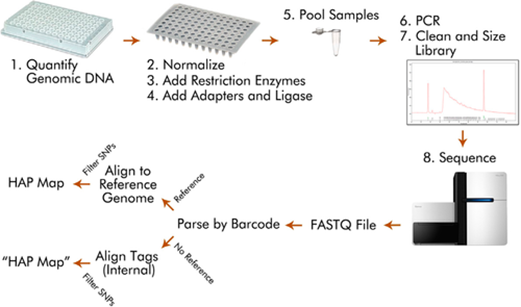
Fig. 2. Rappresentazione schematica della tecnica GBS (affine al ddRAD). Da Poland e Rife, “Genotyping‐by‐Sequencing for Plant Breeding and Genetics”, The Plant Genome, 2012.
E’ previsto che l’utilizzo della medesima tecnologia in entrambe le specie oggetto del progetto permetterà una maggiore efficienza in termini di costi e di analisi dei dati.
Nel corso della seconda annualità del progetto lo svolgimento delle attività è stato segnato dall’emergenza COVID-19. A partire dai primi mesi del 2020, la pandemia ha avuto effetti marcatamente negativi sulle attività di ricerca in essere da parte di SSSA. In particolare, nella prima metà dell’anno l’accesso ai laboratori è stato interdetto (durante il primo lockdown) e subito dopo contingentato per rispettare le norme in merito al distanziamento personale e alla circolazione. Il COVID-19 ha avuto impatti anche nelle procedure amministrative legate al progetto, e in particolare relativamente all’assunzione di personale. Ciononostante, e grazie al supporto delle unità amministrative SSSA, è stato possibile coinvolgere un’assegnista di ricerca (Mercy Macharia) che ha preso servizio sul progetto a partire dal 01/12/2020.
Le attività di ricerca a carico di SSSA sono state volte alla preparazione al sequenziamento e genotipizzazione di accessioni di miglio e mais che avverrà nel terzo anno di progetto. In particolare, le attività hanno riguardato tre punti: i) la selezione del materiale da genotipizzare, ii) l’estrazione del DNA, e iii) la costruzione di una piattaforma bioinformatica necessaria all’analisi dei dati ed alla produzione dei risultati attesi.
Il primo punto relativo alla selezione del germoplasma ha richiesto la stretta collaborazione con l’unità di ricerca dell’Università degli Studi di Firenze che si è occupata della valutazione in pieno campo di varietà di miglio e di mais. In seguito al confronto dei risultati dell’analisi fenotipica, e seguendo un criterio di massimizzazione del valore potenziale per l’agricoltura toscana delle accessioni da caratterizzare, abbiamo incluso 89 varietà di miglio e 61 varietà di mais, per un totale di 150 genotipi, nell’elenco dei materiali genetici da caratterizzare.
All’attività di selezione del materiale è seguita l’estrazione del DNA genomico, che si è svolta presso i laboratori BioLabs dell’Istituto di Scienze della Vita di SSSA. Per l’estrazione sono stati utilizzati Kit SIGMA GenElute Plant MiniPrep. Brevemente, I campioni sono stati germinati tramite “rolled towel”, ovvero imbibendo i semi in rotoli di carta assorbente ed incubandoli al buio a temperatura costante di 25°C per 6-10 giorni (dipendentemente dal tempo di germinazione delle diverse accessioni). In Figura 1 è mostrata la preparazione di un rolled towel per campioni di mais, prima dell’incubazione. I germogli sono stati congelati in azoto liquido e macinati. L’estrazione si è svolta tramiti lisi cellulare e conseguente precipitazione dei frammenti cellulari per separare gli acidi nucleici da tutte le altre componenti del nucleo e del citoplasma, portandoli in soluzione acquosa. A questo step è seguita la ligazione del DNA ai filtri a base silicea che sono parte del Kit di estrazione ed infine alla sua purificazione ed eluizione in soluzione buffer. Al termine di questo processo, il DNA di ogni singola accessione era disponibile in purezza ed in soluzione acquosa, pronto per essere utilizzato per la produzione di library di sequenziamento (attività del terzo anno di progetto).

Fig. 3. Rolled-towel di mais in preparazione. In ciascun rotolo di carta sono posti a germinare tra i 5 e i 10 semi di un’accessione. Dopo la germinazione, il materiale vegetale viene raccolto e mantenuto a -80°C prima di procedere all’estrazione del DNA.
L’estrazione di DNA ha mostrato sufficiente qualità e quantità del DNA tramite osservazione allo spettrofotometro ed al gel di agarosio (Fig. 2). Per tutte le accessioni di entrambe le specie, il DNA aveva concentrazione superiore ai 13 ng/uL e rapporti di assorbanza 260/280 e 260/230 vicini all’1.00. Il DNA così ottenuto è stato piastrato ed è pronto ad essere sequenziato ed analizzato nel terzo anno del progetto.

Fig. 4. Gel di agarosio che mostra la quantificazione del DNA estratto da un sottoinsieme di campioni coinvolti nello studio. La quantificazione di ciascun avviene in corsie organizzate in tre livelli (dall’alto verso il basso) e in relazione allo standard di concentrazione rappresentato dalle prime tre corsie di ogni fila (λDNA). I campioni presentano buona quantità e qualità del DNA. I numeri campione mostrati nell’immagine, indicati in corrispondenza delle corsie di ciascun campione, derivano dalle pratiche in essere al laboratorio e non hanno attinenza con il reale codice campione usato per il progetto
La terza attività ha riguardato il potenziamento delle capacità informatiche in essere al gruppo SSSA in previsione della disponibilità dei dati genomici nel terzo anno di progetto. In seguito alla situazione della pandemia COVID-19, si è reso necessario lavorare da remoto ed aumentare la quota di lavoro agile necessaria allo svolgimento delle attività progettuali. Per venire incontro a questa evenienza, nonché per impiegare al meglio il tempo di progetto durante la pandemia, si è deciso di anticipare la messa a punto dell’infrastruttura bioinformatica necessaria all’analisi dei dati genomici utilizzando un servizio di noleggio di materiale informatico. Inoltre, in fase di pianificazione delle attività sperimentali, sono emerse delle esigenze specifiche in termini di potere computazionale da spendere nell’analisi dei dati: dalle moderne tecniche di sequenziamento genomico che andremo a utilizzare ci aspettiamo infatti di ottenere almeno 15,000 marcatori molecolari per campione, per un totale di 2,250,000 dati molecolari. Per gestire tali dati è richiesta una buona disponibilità di RAM (almeno 16 GB) e dischi a stato solido (SSD) per velocizzare i processi computazionali. Gli strumenti bioinformatici usati nel progetto sono quindi due computer portatili ed un computer fisso con caratteristiche hardware sufficienti a supportare le analisi richieste dal progetto. In seguito alla disponibilità del materiale informatico è stato possibile installare tutti i programmi necessari all’analisi dei dati e verificarne la funzionalità tramite dataset dummy, ovvero dei dati fittizi ma della forma e qualità dei dati che ci aspettiamo ottenere dal sequenziamento genomico. I programmi utilizzati includono diversi strumenti sviluppati per sistemi Windows e Unix, tra i quali Tassel (https://www.maizegenetics.net/tassel) Structure https://web.stanford.edu/group/pritchardlab/structure.html) e numerosi pacchetti di analisi disponibili per il linguaggio di programmazione R (https://cran.r-project.org/), tra cui il pacchetto R/adegenet (https://adegenet.r-forge.r-project.org/) ed il pacchetto R/GAPIT (http://www.zzlab.net/GAPIT/).
Questi programmi saranno utilizzati per estrarre informazioni derivanti dal sequenziamento, ed in particolare caratterizzare il grado di diversità genetica esistente nelle popolazioni di mais e di miglio analizzate dal progetto. Grazie all’uso di questi approcci, sarà possibile identificare le caratteristiche genetiche e molecolari delle singole accessioni e quindi, in combinazione con le attività di campo svolte da altri partner del progetto, l’individuazione di genotipi con combinazioni genetiche e fenotipiche di interesse.
Elenco prodotti concreti del WP:
Elenco prodotti concreti del WP5:
L’elenco dei prodotti attesi dall’attività SSSA è il seguente:
- Caratterizzazione genetica e molecolare delle singole accessioni
- Individuazione di genotipi con combinazioni genetiche e fenotipiche di interesse
- Avvio di un processo di miglioramento genetico focalizzato alla componente nutritiva, all’adattabilità agli ambienti toscani e alla resilienza ai cambiamenti climatici.
I prodotti saranno disponibili al termine del terzo anno di attività di progetto. In particolare, il prodotto 1 dipende dalle attività di sequenziamento che sono pianificate per i mesi di Gennaio e Febbraio 2021. I prodotti 2 e 3 dipendono dal precedente, e saranno sviluppati una volta che i dati molecolari saranno pronti.
